 A growing body of research is implicating mitochondrial dysfunction as a root cause of a wide spectrum of metabolic, lifestyle and degenerative diseases including: Chronic Fatigue Syndrome, Autism, Cancer, Diabetes, Alzheimer’s, Fibromyalgia and potentially many more. Essential to human survival at the most basic level we review the evidence that these specialized organelles are suffering in our modern environment.
A growing body of research is implicating mitochondrial dysfunction as a root cause of a wide spectrum of metabolic, lifestyle and degenerative diseases including: Chronic Fatigue Syndrome, Autism, Cancer, Diabetes, Alzheimer’s, Fibromyalgia and potentially many more. Essential to human survival at the most basic level we review the evidence that these specialized organelles are suffering in our modern environment.
Mitochondria and Energy Production
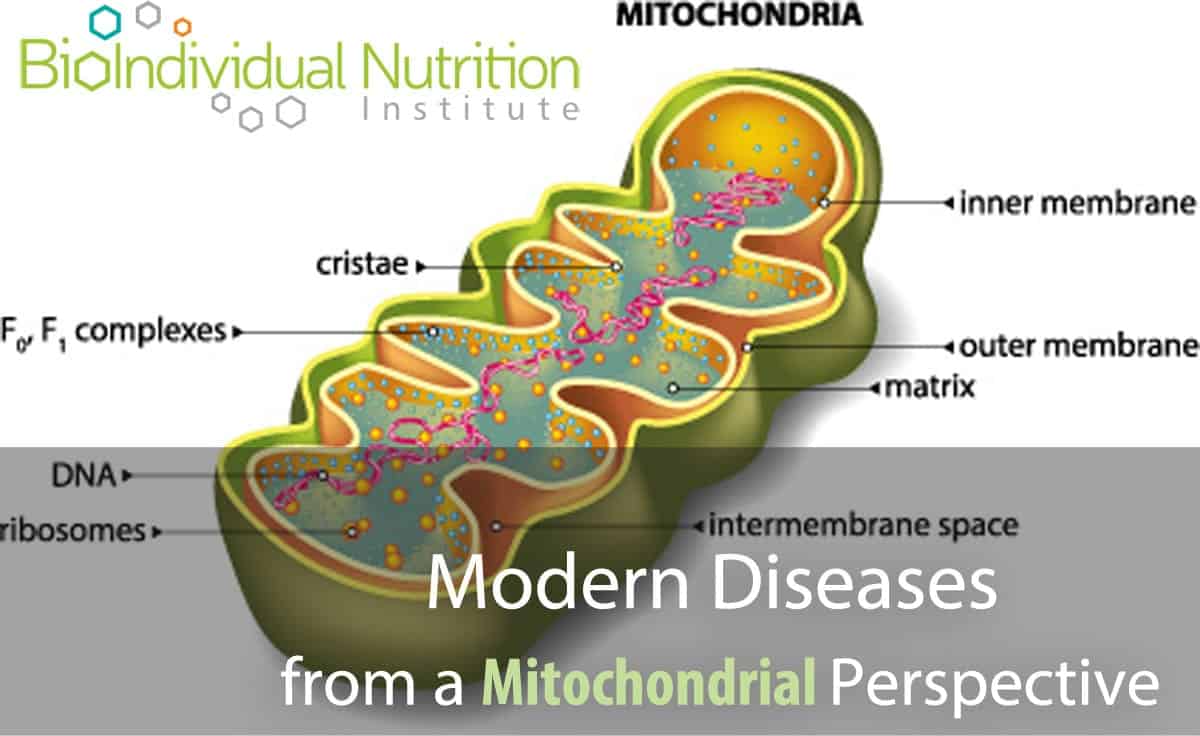
These minute powerhouses in every cell are responsible for regulating and storing cellular energy. They convert ingested energy, for example glucose, into a format the body can actively use, called ATP (Adenosine Tri Phosphate).
ATP carries energy within its atomic bonds which can be released for use in biochemical processes. Hydrolysis of ATP to ADP (Adenosine Di Phosphate) liberates energy: breaking the phosphate bond is exothermic (it gives off energy) due to the inherent instability of ATP which would ‘prefer’ to be in its ADP state. After conversion of ATP to ADP, and release of the energy holding the phosphate in place, the ATP molecule (now ADP) is ‘spent’ and needs to be recycled.
Electrochemical reactions (collectively known as oxidative phosphorylation) take place inside the mitochondria along the electron transport chain to reattach a phosphate group to create ATP again. This endless recycling of ADP to ATP is needed to maintain energy production.
Even though each cell contains approximately one billion ATP molecules it is only sufficient to meet the cell’s energy requirements for a few minutes and constant activity is required to keep energy flowing – amazingly humans manufacture and consume their own weight in ATP every day[1]. Cells needs energy to function optimally so even a minor impairment of mitochondrial function can have dramatic systemic ramifications.
The Scientific Study of Mitochondria
Mitochondria are actually bacterial in nature and they evolved with us endosymbiotically: mitochondrial DNA is inherited through the maternal line. [2] Unable to maintain themselves outside of eukaryotic cells, mitochondria have been co-opted into our energy production systems. However, they have their own genetic system distinct from the nuclear genome – they even use a different protein coding system for translation of mitochondrial DNA.[3]
Mitochondrial DNA is exquisitely sensitive to environmental challenges, specifically oxidative damage and stress, due to the high ratio of coding regions (versus non-coding regions) in the DNA; and a lack of protective histones supporting their DNA structure and environment.[4] Alarmingly, oxidative damage occurs at a rate five to ten times greater in mitochondrial DNA versus nuclear DNA and “damaged mitochondria promptly accelerate intra-cellular oxidation”.[5]
De Novo mutations have been specifically linked to both autism and schizophrenia indicating that the DNA damage leading to the disease is developed and not inherited.[6] Specific alterations in the mitochondrial DNA coding for complexes in the electron transport chain and pyruvate dehydrogenase have been identified in the frontal cortex of patients with autism which cause inadequate (anaerobic) utilization of glucose and an excess of unwanted metabolic byproducts.[7]
Mitochondria uniquely sit in two very different areas of biological research: structural (proteins, tissues, genome etc.) and bioenergetics (energy metabolism). These interfacing areas of research have until relatively recently been uncoupled and in vivo research has been difficult due to a lack of biomarkers and assays to detect mitochondrial damage. This lack of ability to diagnose problems with energy flow in and out of the mitochondria means the root cause of many diseases relating to mitochondrial function has been overlooked or ignored.[8]
Mitochondrial Function and Biogenesis
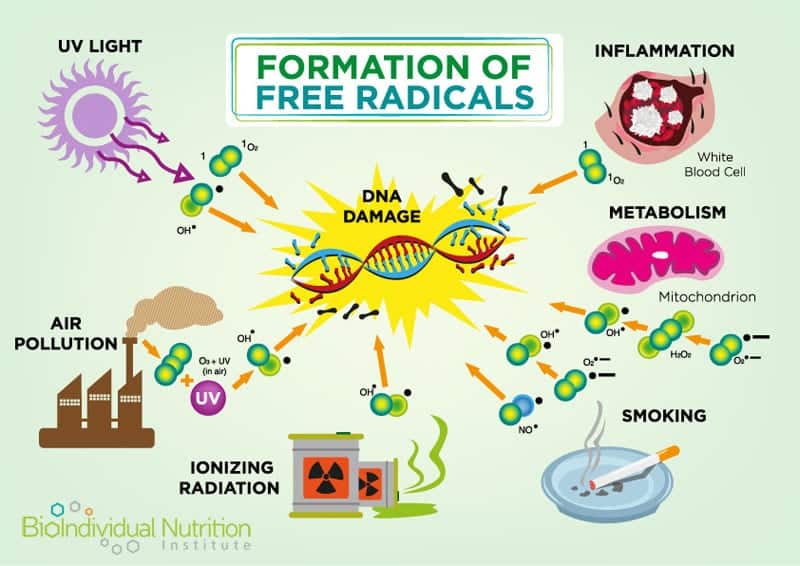 In addition to their role in energy regulation, mitochondria are also involved in the maintenance of intracellular calcium levels and calcium buffering (required for cellular signaling)[9]. They also regulate cell numbers and defend against unwanted or dangerous cells by triggering programmed cell death (apoptosis).[10] Signaling between the human (nuclear) and mitochondrial genome is also controlled by the mitochondria themselves via the production of Reactive Oxygen Species (ROS).
In addition to their role in energy regulation, mitochondria are also involved in the maintenance of intracellular calcium levels and calcium buffering (required for cellular signaling)[9]. They also regulate cell numbers and defend against unwanted or dangerous cells by triggering programmed cell death (apoptosis).[10] Signaling between the human (nuclear) and mitochondrial genome is also controlled by the mitochondria themselves via the production of Reactive Oxygen Species (ROS).
Excessive ROS (also known as free radicals) is a metabolic byproduct and one of the triggers for apoptosis. For example, asbestos induced lung disease has been linked to elevated alveolar epithelial apoptosis due to increased ROS caused by the asbestos damage.[11] Elevated levels of mitochondrial calcium can also trigger apoptosis and recent research indicates that apoptosis of nerve cells contributes to Alzheimer’s.[12]
While mitochondria have their own unique DNA, the majority of the proteins required to synthesize the organelle are recruited from nuclear coded proteins. This means they need to communicate effectively with nucleic DNA to produce the protein pieces required to join with existing subcomponents. Since the co-ordination required for mitochondrial biogenesis requires the expression of two different genomes, any epigenetic problems with DNA signaling due to a lack of nutrients or toxicity are amplified.[13]
Pharmacology and Mitochondrial Disease
Azidothyramidine was commercialized for the treatment of HIV/AIDs in the late 1980’s and transformed the disease from a death sentence to a chronic manageable condition. Unfortunately, prolonged treatment with the drug, and others nucleoside analogs, has severe or fatal side effects. Over 30% of patients treated experienced heart muscle damage and loss of vision with mitochondrial toxicity (via multiple routes) being the agreed cause.[14]
The association with pharmacologically active compounds and mitochondrial function is now so well recognized that pharmaceutical companies have developed high throughput screening to specifically look for mitochondrial damage during the drug development process. Other classes of compounds including antibiotic, antiepileptic, antidiabetic, antipsychotic, antidepressant, beta-blocker, and nonsteroidal anti-inflammatory drugs have also demonstrated the ability to damage mitochondria; many others may reveal the same problems with further research.[15]
Fueling the Mitochondria
Mitochondria can use different sources of fuel (glucose, protein and fat) both with and without oxygen depending on the cellular conditions. A delicate balance of regulatory mechanisms and hormones determines which of these fuel sources should be preferentially utilized. The byproducts of the different pathways also control a wide variety of downstream biological processes meaning that fuel availability (or capacity to utilize it) has wider implications.
Mitochondrial fuel selection, and metabolite production, is interlinked with responses in vital systems such as immune function[16]. For example an excessive production of ROS stimulates the inflammatory response[17] and can trigger chronic diseases.[18]
Energy Preferences
Organs have different metabolic requirements: the brain relies on glucose and a derivative of fatty acids called ketone bodies while muscle uses glucose for short bursts of energy but uses fatty acids for 85% of its needs. The heart relies exclusively on anaerobic metabolism of fatty acids and has a high density of mitochondria to meets its energy requirements. [19]
Aerobic glycolysis of glucose (in the presence of oxygen) is 5 times more efficient than anaerobic glycolysis as it fully breaks down the glucose. The buildup of lactic acid, as a result of anaerobic glycolysis, can be used to indicate a drop in mitochondrial performance as it no longer metabolizes the pyruvate to release the remaining energy – elevated lactate to pyruvate ratios are often seen in patients with autism.[20] When mitochondria are utilizing fuel less efficiently they produce more ROS (like a sooty fire) and this causes further damage to the delicate organelle.
Dietary modifications to reduce glucose have been found to improve some behavioral traits commonly associated with autism in mice models. Changes in plasma metabolites reduce the neuroinflammation and impaired neurogenesis seen in both animal and human models. [21]
The excess ROS production (due to inefficient fuel usage), combined with low levels of intrinsic antioxidants and lack of precursors needed for fatty acid metabolism (specifically carnitine) often associated with autism explains why some patients experience improved symptoms with a ketogenic (fat based) diet especially when combined with carnitine supplementation. [22]
Mitochondrial Diseases and Dysfunctions
There are an increasing number of diseases which have been attributed to mitochondrial function and many share general symptoms of fatigue and muscle pain due to impaired energy provision to organs with high energy demands.
At the other end of the disease spectrum, the fatal condition MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke like episodes) causes extreme seizures, migraines and brain damage. Mutations in mitochondrial DNA mean the body is not able to meet its own energy needs, specifically during physical exertion.
While some mitochondrial diseases have a strong genetic element many are suspected to be as a result of environmental conditions which do not support efficient energy utilization. Mitochondrial dysfunction as a result of increased ROS production, lack of antioxidants and environmental pollutants has been coming under closer scrutiny as a trigger or even for conditions as diverse as autism and Gulf War Syndrome.[23] Another metabolic waste product, oxalates cause oxidative stress too and further damage the mitochondria leading to chronic disease.
The high energy demands of the brain make it more susceptible to a buildup of ROS if mitochondria are not functioning optimally. Additionally, the ROS directly damage the polyunsaturated fats which make up the majority of the brain tissue and mitochondrial damage has been implicated in a variety of neurodegenerative diseases including Alzheimer’s.[24]
Chronic stress has also been shown to inhibit mitochondrial function and induces reversible mitochondrial damage causing symptoms resembling Irritable Bowel Syndrome (IBS).[25] Sleep apnea is another condition which reduces mitochondrial function by causing frequent periods of hypoxia.[26] Glutamate has also been found to contribute to mitochondrial disease because it interferes with calcium homeostasis.[27]
Autistic Spectrum Disorder
The spectrum of symptoms, and wide variety in the severity of autism spectrum disorder, supports the notion that varying energy demands and fuel utilization throughout the body determine the degree of the impact from poorly functioning mitochondria. The huge degree of divergence points to multiple genetic susceptibilities which have been found to cluster around genes responsible for calcium and metabolic (MAPK) signaling. These foundational errors also indicate widespread “vulnerability to other chronic and systemic problems potentially including cancer, metabolic conditions and heart diseases” for both autistic patients and the public at large.[28]
Neurological symptoms are virtually always present and this is the organ with the highest energy usage and very stringent requirements about fuel sources (other high energy systems such as the bowels and muscles are also typically symptomatic).
Some patients also experience idiopathic symptoms including cardiovascular symptoms, growth retardation and fatigue which align with the known outcomes of poor mitochondrial function. [29] According to one study, defects in oxidative phosphorylation (the process by which ADP is remade into ATP) are identified in nearly 50% of patients with autism highlighting the need for metabolic evaluation to support individualized therapy.[30] Glutamate levels are also often elevated in ASD patients, further exacerbating mitochondrial toxicity.[31]
Cardiovascular Health
The heart is another organ which has high energy demands and 35% of the volume of cardiac muscles cells is taken up by mitochondria. Diabetic patients often experience cardiac problems higher than predicted levels (adjusting for hypertension and coronary artery disease) due to deficiencies in cardiac energy metabolism.[32] Various mechanisms have been identified which contribute to the multifactorial cardiac challenges, including: increased ROS production; impaired calcium regulation and incomplete biogenesis of new mitochondria.
The root cause of cardiovascular diseases, including atherosclerosis, is increasingly being linked to the mitochondria responsible for energy production and regulation. The vicious feedback loop of oxidative stress causing inflammatory responses and apoptosis further exacerbates a system under pressure leading to a variety of disease states.[33]
Cancer and Oncogenesis
Since the beginning of the 20th century, scientists knew that cancer cells had differences in their metabolism: preferentially utilizing glucose anaerobically and producing a lot of lactic acid (known as the Warburg effect) even when sufficient oxygen is available for aerobic metabolism. It is postulated that anaerobic glycolysis actually produces more of the biosynthetic intermediates needed for cellular growth and proliferation.[34]
Abnormal mitochondrial function has been found in multiple human cancer variants. In vitro replication of mitochondrial mutations, knocking out specific parts of the electron transport chain, has demonstrated cells with increased ROS (as a result of anaerobic glycolysis) have higher invasive behaviors and greater migration rates comparable to cancer cells.[35]
Additionally, some metabolic enzymes typically involved in the Krebs cycle usually actually act as oncosuppressors – this means that imbalances in fuel selection due to mitochondrial dysfunction also impacts genes promoting cell proliferation. [36]
The regulatory role of mitochondria in apoptosis is also under scrutiny for its role in cancer progression as compromised apoptotic function removes the usual protection the body has for removing dangerous cells. Cancerous cells actually manage to deregulate the pathway which would typically save the rest of the body from tumor development.
Elevated ROS, from poorly functioning mitochondria, also increases cancer risk by damaging the DNA and stimulating pro-growth responses (tumor genesis). In an attempt to slow down the tumorigenic signaling, the body will typically respond by up-regulating the production of intrinsic anti-oxidants to reduce ROS levels.
The relationship between ROS, antioxidants and tumor genesis explains why research has repeatedly substantiated claims that fresh fruits and vegetables, packed with anti-oxidants, can prevent and even cure cancers and nutritional therapies reduce ROS while providing the nutrients needed for the body to heal itself.[37]
Evidence for other Mitochondrial Conditions
Metabolic Syndrome: Many of the risk factors for metabolic syndrome (obesity, elevated cholesterol, hypertension and diabetes) are linked with abnormal mitochondrial function. [38] Over nutrition is specifically linked with oxidative stress especially when combined with a lack of exercise.[39]
Asthma: Animal models have demonstrated the role of oxidative stress in the inflammatory response triggered during asthma. [40] Additionally, there is an increased likelihood of severe asthma attacks in obese individuals, pointing to mitochondria as the root cause of the disease.[41] The use of targeted antioxidant therapy in animal asthma models has already demonstrated a reduction in the fibrotic airway remodeling which is linked to disease progression.[42]
Chronic Fatigue Syndrome: The severity of symptoms presented by sufferers of CFS has been directly linked to the degree of mitochondrial disease. [43]
Fibromyalgia: The chronic pain of this condition has been linked to suboptimal mitochondrial function in the nervous system and patients are often low in enzymes required for complete energy metabolism, namely CoQ10.[44]
Irritable bowel syndrome: Mitochondrial DNA mutations have been found to be higher in patients with IBS and oxidative stress triggers damaging chronic inflammatory responses.[45]
Diabetes: Excessive production of ROS contribute to the pathophysiology of diabetes, specifically damaging nerves and the eyes.[46]
There is increasing evidence that mitochondrial dysfunction is responsible for, or strongly contributes to, many more chronic degenerative disorders, including: Alzheimer’s, Parkinson’s disease, Huntington Disease, Amyotrophic Lateral Sclerosis (ALS) oobesity and autoimmune diseases (lupus, Sjogren’s syndrome and rheumatoid arthritis).[47]
The New Paradigm
Science tends to put things in little, separate, boxes. Taking a step back and looking at the wider picture we can see that the various epidemics of modern disease present very different symptoms but the root cause could be remarkably simple.
Putting together the pieces of the puzzle, we can see that our ancient mitochondria can simply not cope with the insult of environmental toxins, from the food we eat; medicines we take; air we breathe; and chemicals we apply on our body. Most likely the lack of quality nutrients in the modern processed diet is also responsible for the reduced function and increasing mitochondrial damage.
Unless some drastic action is taken to improve the working conditions of these vital organelles, the progression and severity of lifestyle diseases is set to continue at an alarming rate. Figures for diabetes, cancers, cardiovascular disease and more are soaring and the abuse of our mitochondria seems to be the elephant in the room that has finally been noticed.
References
[1] Charles W Schmidt, “Unraveling Environmental Effects on Mitochondria”, Environ Health Perspectives 2010 Jul; 118(7)A-292-297. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2920932/
[2] Michael W Gray, Gertraud Burger, and B Franz Lang, “The Origin and Early Evolution of Mitochondria”, Genome Biology 2001 June 5; 2(6): reviews 1018.1-reviews1018.5 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC138944/
[3] Cooper GM, The Cell: A Molecular Approach, 2nd Edition. Sunderland (MA): Sinauer Associates; 2000. Mitochondria Available from: https://www.ncbi.nlm.nih.gov/books/NBK9896/
[4] Charles W Schmidt, “Unraveling Environmental Effects on Mitochondria”, Environ Health Perspectives 2010 Jul; 118(7)A-292-297, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2920932/
[5] Harvard School of Public Health, Baccarelli Lab, Mission, Mitochondriomics
https://www.hsph.harvard.edu/baccarelli-lab/mission/environmental-mitochondriomics/
[6] “Autism and Schizophrenia: Scientists Measure Gene Mutation Rate”, September 3rd, 2010, https://research.chusj.org/en/Communications/Nouvelles/2010/Autisme-et-schizophrenie-Des-chercheurs-evaluent-l
[7] Gu, F., Chauhan, V., Kaur, K., Brown, W. T., LaFauci, G., Wegiel, J., & Chauhan, A. (2013). “Alterations in mitochondrial DNA copy number and the activities of electron transport chain complexes and pyruvate dehydrogenase in the frontal cortex from subjects with autism” Translational Psychiatry, 3(9), e299, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3784762/
[8] Charles W Schmidt, “Unraveling Environmental Effects on Mitochondria”, Environ Health Perspectives 2010 Jul; 118(7)A-292-297. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2920932/
[9] Rosario Rizzuto, Diego De Stefani, Anna Raffaello & Cristina Mammucari “Mitochondria as sensors and regulators of calcium signaling” Nature Reviews Molecular Cell Biology 13, 566-578 (September 2012) https://www.nature.com/nrm/journal/v13/n9/full/nrm3412.html
[10] Jerome Estaquier, Francois Vallette, Jean-Luc Vayssiere, and Bernard Mignotte, Advances in Experimental Medicine and Biology, 2011 December 22; 942:157-183, https://www.ncbi.nlm.nih.gov/pubmed/22399422
[11] Kamp DW, “Asbestos-Induced Lung Diseases: An Update”, Translational Research The Journal of Laboratory and Clinical Medicine, 2009 April, 153(4):143-152, https://www.ncbi.nlm.nih.gov/pubmed/19304273
[12] Qi H, Shuai J, “Alzheimer’s Disease via Enhanced Calcium Signaling Caused by the Decrease of Endoplasmic Reticulum-Mitochondrial Distance”, Med Hypotheses 2016 April;89:28-31 https://www.ncbi.nlm.nih.gov/pubmed/26968904
[13] Ryan MT and Hoogenraad NJ, “Mitochondrial-Nuclear Communications”, Annual Review of Biochemistry, 2007 July, Volume 76:701-722, First Published as a Review in Advance on January 16, 2007, https://www.ncbi.nlm.nih.gov/pubmed/17227225
[14]Erin R Scruggs and Amie J Dirks Naylor, “Mechanisms of Zidovudine-Induced Mitochondrial Toxicity and Myopathy”, Pharmacology 2008;82-83-88, https://www.karger.com/Article/Pdf/134943
[15] Neustadt J and Pieczenik SR, “Medication-Induced Mitochondrial Damage and Disease”, Molecular Nutrition and Food Research, 2008 July;52(7):780-788, https://www.ncbi.nlm.nih.gov/pubmed/18626887
Mitoaction, Table of Reported Drugs with Mitochondrial Toxicity https://www.mitoaction.org/files/MitoToxins_0.pdf
Garrecht M and Austin DW, “The Plausibility of a Role For Mercury in the Etiology of Autism: A Cellular Perspective”, Toxicological and Environmental Chemistry, 2011 May-July; 93(5-6):1251-1273, https://www.ncbi.nlm.nih.gov/pubmed/22163375
[16] Agilent Technologies, “Mitochondria Fuel Flexibility, Dependency & Capacity and the Seahorse XF Mito Fuel Flex Test”, https://www.agilent.com/en-us/products/cell-analysis-(seahorse)/mitochondrial-fuel-flexibility-the-xf-mito-fuel-flex-test
[17] MS Williams and J. Kwon, “T Cell Receptor Stimulation, Reactive Oxygen Species, and Cell Signaling”, Free Radical Biology Med. 2004 Oct 15;37(8):1144-51, https://www.ncbi.nlm.nih.gov/pubmed/15451054
[18] Lobo, V., Patil, A., Phatak, A., & Chandra, N., “Free Radicals, Antioxidants and Functional Foods: Impact on Human Health, Pharmacognosy Reviews, 2010 Jul-Dec; 4(8), 118–126. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3249911/
[19] Berg JM, Tymoczko JL, Stryer L. Biochemistry, “Each Organ Has a Unique Metabolic Profile”, 5th edition. New York: W H Freeman; 2002. Section 30.2, Each Organ Has a Unique Metabolic Profile. https://www.ncbi.nlm.nih.gov/books/NBK22436/
[20] J Jay Gargus Department of Physiology and Biophysics and Department of Pediatrics, Section of Human Genetics/School of Medicine & Faiqa Imtiaz, Arabian Diagnostics Laboratory, King Faisal Specialist Hospital and Research Centre, “Mitochondrial Energy-Deficient Endophenotype in Autism”, American Journal of Biochemistry and Biotechnology 4(2):198-207 2008, https://thescipub.com/PDF/ajbbsp.2008.198.207.pdf
[21] A Currais, C Farrokhi, R Dargusch, M Goujon-Svrzic, and P Maher, “Dietary Glycemic Index Modulates The Behavioral and Biochemical Abnormalities Associated with Autism Spectrum Disorder”, Molecular Psychiatry 21; 426-436, Published online June 9, 2015, https://www.nature.com/mp/journal/v21/n3/abs/mp201564a.html
[22] Eleonora Napoli, Nadia Duenas, Cecilia Giulivi, “Potential Therapeutic Use of Ketogenic Diet in Autism Spectrum Disorders”, Frontiers in Pediatrics, 2014 June 30; 2:69, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4074854/
[23] Beatrice Golomb MD, PhD, “Oxidative Stress and Mitochondrial Injury in Chronic Multisymptom Conditions: From Gulf War Illness to Autism Spectrum Disorder”, Nature Proceedings : hdl:1010/npre.2012.6847.1: January 30, 2012, https://drmyhill.co.uk/drmyhill/images/d/df/Oxidative_Stress_and_Mitochondrial_Injury_in_CMC.pdf
[24] Barry Halliwell, “Reactive Oxygen Species and the Central Nervous System” , Journal of Neurochemistry, Volume 59;(5) 1609-1623, November 1992, https://www.ncbi.nlm.nih.gov/pubmed/1402908
[25] Vicario M, Alonso C, Guilarte M, Serra J, Martinez C, Gonzalez-Castro AM, Lobo B, Antolin M, Andreu AL, Garcia-Arumi E, Casellas M, Saperas E, Malagelada JR, Azpiroz F, Santos J. “Chronic Psychosocial Stress Induces Reversible Mitochondrial Damage and Corticotropin-Releasing Factor Receptor Type-1 Upregulation In The Rat Intestine and IBS-Like Gut Dysfunction”, Psychoneuroendocrinology 2012 January;37(1):65-77, https://www.ncbi.nlm.nih.gov/pubmed/21641728
[26] Yan Deng, Xue-Ling Guo, Xiao Yuan, Jin Shang, Die Zhu, and Hui-Guo Liu, “P2X7 Receptor Antagonism Attenuates The Intermittent Hypoxia-Induced Spatial Deficits In A Murine Model Of Sleep Apnea Via Inhibiting Neuroinflammation and Oxidative Stress”, Chinese Medical Journal (English) 2015 August 20; 128(16): 2168-2175, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4717977/
[27] Cláudia F Pereira , Catarina Resende de Oliveira, “Oxidative glutamate toxicity involves mitochondrial dysfunction and perturbation of intracellular Ca2+ homeostasis”, Neuroscience Research, Volume 37, Issue 3, July 2000, Pages 227–236, https://www.sciencedirect.com/science/article/pii/S0168010200001243
[28] Wen Y, Alshikho MJ, Herbert MR, “Pathway Network Analyses for Autism Reveal Multisystem Involvement, Major Overlaps with Other Diseases and Convergence upon MAPK and Calcium Signaling.”, PLoS One. 2016 Apr 7;11(4):e0153329, https://www.ncbi.nlm.nih.gov/pubmed/27055244.
[29] Jacqueline R Weissman, Richard I Kelley, Margaret L Bauman, Bruce H Cohen, Katherine F Murray, Rebecca L Mitchell, Rebecca L Kern, and Marvin R Natowicz, “Mitochondrial Disease In Autism Spectrum Disorder Patients: A Cohort Analysis”, PLos ONE 3(11): e3815 November 26, 2008, https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0003815.
[30] Poling JS, Frye RE, Shoffner J, Zimmerman AW, “Developmental Regression and Mitochondrial Dysfunction in a Child With Autism” J Child Neurol.2006; 21(2):170-172. https://www.ncbi.nlm.nih.gov/pubmed/16566887
[31] Atsuko Shinohea, Kenji Hashimotob, , , Kazuhiko Nakamuraa, Masatsugu Tsujiic, Yasuhide Iwataa et al, “Increased serum levels of glutamate in adult patients with autism”, Progress in Neuro-Psychopharmacology and Biological Psychiatry, Volume 30, Issue 8, 30 December 2006, Pages 1472–1477, https://www.sciencedirect.com/science/article/pii/S0278584606002697.
[32] Bugger H and Abel ED, “Mitochondria In The Diabetic Heart”, Cardiovascular Research, 2010 Nov 1;88(2): 229-240, https://www.ncbi.nlm.nih.gov/pubmed/20639213
[33] Ballinger SW, “Mitochondrial Dysfunction in Cardiovascular Disease”, Fee Radical Biol Med. 2005 May 15;38(10):1278-95, https://www.ncbi.nlm.nih.gov/pubmed/15855047
[34] Fogg VC, Lanning NJ, and Mackeigan JP, “Mitochondria in Cancer: At The Crossroads of Life and Death”, Chinese Journal of Cancer, 2011 Aug;30(8):526-539, https://www.ncbi.nlm.nih.gov/pubmed/21801601
[35] Jia Ma, Qing Zhang, Sulian Chen, Binbin Fang, Qingling Yang, Changje Chen, Lucio Miele, Fazlul H Sarkar, Jun Xia, and Zhiwei Wang, “Mitochondrial Dysfunction Promotes Breast Cancer Cell Migration and Invasion through HIF1a Accumulation via Increased Production of Reactive Oxygen Species”, Plos ONE, July 29, 2013, https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0069485
[36] Scatena R, “Mitochondria and Cancer: A Growing Role In Apoptosis, Cancer Cell, Metabolism, and Dedifferentiation”, Advances in Experimental Medicine and Biology, 2011 December 22, Ch. Advances in Mitochondrial Medicine, Volume 942; 287-308, https://www.ncbi.nlm.nih.gov/pubmed/22399428
[37] Gerson Institute, https://gerson.org/gerpress/
[38] Ulaganathan Mabalirajan and Balaram Ghosh, “Mitochondrial Dysfunction in Metabolic Syndrome and Asthma”, Journal of Allergy, 2013 June 5, 340476, https://europepmc.org/articles/PMC3687506/
[39] James AM, Collins Y Logan A, and Murphy MP, “Mitochondrial Oxidative Stress and the Metabolic Syndrome”, Trends in Endocrinology & Metabolism, September 2012 Volume 23(9);429-434, https://www.ncbi.nlm.nih.gov/pubmed/22831852
[40] P Hemachandra Reddy, “Mitochondrial Dysfunction and Oxidative Stress in Asthma: Implications for Mitochondria Targeted Antioxidant Therapeutics”, Pharmaceuticals (Basel) 2011Mar. 4(3):429-456, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3066010/
[41] Ulaganathan Mabalirajan and Balaram Ghosh, “Mitochondrial Dysfunction in Metabolic Syndrome and Asthma”, Journal of Allergy, 2013 June 5, 340476, https://europepmc.org/articles/PMC3687506/
[42] Jaffer OA1, Carter AB, Sanders PN, Dibbern ME, Winters CJ, Murthy S, Ryan AJ, Rokita AG, Prasad AM, Zabner J, Kline JN, Grumbach IM, and Anderson ME, “Mitochondrial-Targeted Antioxidant Therapy Decreases Transforming Growth Factor-β-Mediated Collagen Production In a Murine Asthma Model”, American Journal Of Respiratory Cell and Molecular Biology, 2015 January;52(1):106-15, https://www.ncbi.nlm.nih.gov/pubmed/24988374
[43] Sarah Myhill, Norman E. Booth, and John McLaren-Howard, “Chronic Fatigue Syndrome and Mitochondrial Dysfunction” International Journal of Clinical Experimental Medicine, 2009, 2(1):1-16, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2680051/
[44] Cordero MD, de Miguel M, Carmona-Lopez, Campa F, and Moreno-Fernandez AM, “Oxidative Stress and Mitochondrial Dysfunction in Fibromyalgia” Neuro Endocrinlogy Lett. 2010; 31(2):169-73, https://www.ncbi.nlm.nih.gov/pubmed/20424583
[45] Wei-Feng Wang, Xin Li, Ming-Zhou Guo, Jian-De Chen, Yun-Sheng Yang, Li-Hua Peng, Yong-Hua Wang, Chun-Yan Zhang, and Hui-Hui Li, “Mitochondrial ATP 6 and * Polymorphisms in Irritable Bowel Syndrome with Diarrhea”, World Journal of Gastroenterology, 2013 June 28; 19(24): 3847-3853 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3699044/
Asima Bhattacharyya, Ranajoy Chattopadhyay, and Sheila E Crowe, “Oxidative Stress: An Essential Factor in the Pathogenesis of Gastrointestinal Mucosal Diseases”, Physiological Reviews 2014 April: 94(2):329-354, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4044300/
[46] William I Sivitz, and Mark A Yorek, “Mitochondrial Dysfunction in Diabetes: From Molecular Mechanisms to Functional Significance and Therapeutic Opportunities”, Antioxidants & Redox Signaling, 2010 February 15; 12(4):537-577, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2824521/
[47] United Mitochondrial Disease Fundation, Understanding Mitochondrial Disease, https://www.umdf.org/site/pp.aspx?c=8qKOJ0MvF7LUG&b=7934637

